This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
What You Should Know: – Nonin Medical , a global leader in noninvasive medical monitoring lauches TruO2 OTC, the first over-the-counter (OTC) fingertip pulse oximeter to receive FDA clearance. – Nonin Medical’s founder, Phil Isaacson, was the original inventor of the fingertip pulse oximeter.
Food and Drug Administration (FDA), in fulfilling its task of ensuring that drugs are safe and effective, has recently turned its attention to the growing use of artificial intelligence (AI) and machine learning (ML) in drug development. By Matthew Chun The U.S. manufacturing process design), implement advanced process controls (e.g.,
Recently cleared by the FDA, Osteoboost is clinically proven to significantly slow bone density and strength loss through its patented precision vibration therapy, specifically targeting postmenopausal women with osteopenia.
Food and Drug Administration (FDA) for post-traumatic stress disorder (PTSD) means the drug may be rescheduled, which will lead to substantially decreased regulations attached to it. However, in 2016, DEA added some clarity to the “accepted medical use” qualification, saying that FDA approval was one way to achieve this threshold.
TytoCare lung exam What You Should Know: – TytoCare , a pioneering virtual care company dedicated to delivering accessible, high-quality primary care from the comfort of home announced the company has become the first in the world to receive FDA clearance for its AI-based detection of all three major abnormal lung sounds.
– As the first FDA-cleared self-neuromodulation device for post-traumatic stress disorder (PTSD), Prism for PTSD™ offers a new approach to managing this debilitating condition.
But the FDA has learned some unauthorized versions of their diagnostics have entered the country. The manufacturers have all received emergency use authorizations for antigen tests.
Bakul Patel, the Center for Devices and Radiological Health's chief digital health officer, will help FDA meet its 2022-2025 strategic priorities and set regulatory policies for the new technologies such as AI and machine learning.
Food and Drug Administration (FDA) holding them accountable. Researchers, vendors, and organizations who dont uphold these standards or use data that lacks integrity could face debarment from future clinical trials by the FDA. What Lands Someone on the FDA Debarment List for Clinical Trials?
"Any place that gets cut, it's going to have an impact, because there's not any spare personnel at FDA,” said former agency commissioner Robert Califf, of the layoffs.
Food and Drug Administration (FDA) has approved the Teal Wand , marking it as the first and only at-home self-collection device for cervical cancer screening in the United States. The Teal Wand simply offers a different, FDA-approved method for collecting the sample.
– As the first and only FDA-cleared device of its kind for low bone density, Osteoboost introduces a novel, protective, and preventative approach to bone health, offering a new option for tens of millions of individuals currently without effective interventions. – The launch comes as societal views on aging are evolving.
The draft guidance, which replaces a 2018 document, sets recommendations for how medical device companies should approach cybersecurity in premarket submissions.
The clearance is a major shift for the agency, which described its decision as a “first step” in allowing Florida to bulk purchase lower-cost Canadian medicines.
Food and Drug Administration (FDA) banned the use of Red Dye No. Food and Drug Administration (FDA) banned the use of Red Dye No. The Regulatory Framework for Food and Color Additives FDA regulates the U.S. While a seemingly innocuous phrase, this language has come to haunt FDA. 15, 2025, the U.S. 15, 2025, the U.S.
– The FDA granted a De Novo Classification Request for CT-132, signifying the novel nature of this digital intervention. This strategy aligns with the increasing interest and FDA draft guidance on Prescription Drug Use-Related Software (PDURS). Safety Information There are no contraindications to using CT-132.
Scott Gottlieb supports a reversion to an earlier interpretation of the 21st Century Cures Act, which would exempt more types of clinical decision support software from the FDA’s premarket review process.
The agency’s approval comes months after a large clinical trial showed the drug, called Leqembi, could slow the disease’s progression. Yet experts have raised concerns about its safety.
An FDA announcement that semaglutide injection products, the GLP-1 medication in the blockbuster drugs Wegovy and Ozempic, are no longer in shortage. An FDA announcement that semaglutide injection products, the GLP-1 medication in the blockbuster drugs Wegovy and Ozempic, are no longer in shortage.
The 37-year veteran helmed the agency’s pandemic response, and was a key decision-maker in controversial calls on opioids, an Alzheimer's therapy and muscular dystrophy drugs.
Food and Drug Administration (FDA) regulates tampons and menstrual cups as Class II medical devices. However, the FDA does not require tampon makers to test their products for PFAS or heavy metal contaminants. However, the FDA does not require tampon makers to test their products for PFAS or heavy metal contaminants.
Experts say the layoffs are already causing issues in drug development — and things may get far worse without critical institutional knowledge at the FDA.
Several groups have been granted additional time to comply with the more than 10-year-old legislation, which requires complete end-to-end supply chain visibility for pharmaceutical products.
An agency alert warned that flaws in PTC's Axeda agent and desktop server, used in devices from several manufacturers, could allow an unauthorized attacker to take full control of the host operating system.
By Patrizia Cavazzoni MD & Peter Marks MD PhD - Through the Hub, we plan to foster a community at the FDA for open dialogue and knowledge sharing to identify new approaches to drug and biologic development and overcome hurdles that have traditionally impeded progress for rare disease treatments.
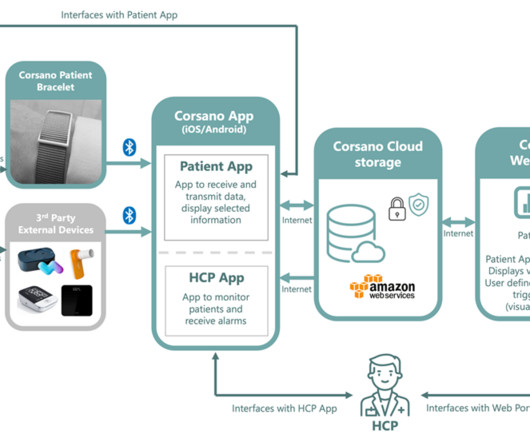
FDA clearance brings one of Europe’s most advanced real-time continuous remote monitoring systems to the US market for the first time. European pedigree FDA clearance is significant for a system which is already CE-MDR medically certified in Europe.
The FDA will hold its first Digital Health Advisory Committee (DHAC) meeting to discuss how the agency should review medical devices that rely on generative AI, like chatbots. |
US Legal Requirements for Quantum-Powered Medical Devices In the United States, some quantum-powered medical devices may be governed under the existing FDA regulatory framework. Foster Institutional Plasticity: Evolve institutions like the FDA and EMA to accommodate quantum technologies.
The HHS Office of Inspector General found that by loosening emergency use authorization requirements to bring COVID-19 tests to market faster, the agency allowed inaccurate tests to be distributed.
Even when manufacturers can develop patches, they may face delays due to FDA regulations that require rigorous testing and approval before updates can be distributed. The Need for FDA Patch Approval Reform The regulatory framework around medical devices creates another major challenge in addressing these vulnerabilities.
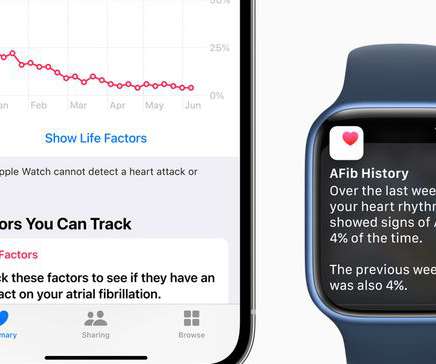
With the new feature, atrial fibrillation patients may have an easier way to track the frequency of the condition over time and see whether lifestyle changes may have positive effects.
Respirators were one of the first medical devices identified as being in critical shortage during the COVID-19 public health emergency, the agency said.
The report proposes multiple actions for protecting older medical devices, including research into more modular devices and collecting data on cyber risks.
The authors list a range of potential positive outcomes of improved device interoperability, using the experience of the consumer technology and telecommunications industries to make their case.
We organize all of the trending information in your field so you don't have to. Join 26,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











































Let's personalize your content