


A Guide to Medical Device Labeling Requirements

The safe operation of a medical device must never come down to guesswork or trial-and-error. Informative, accurate labeling is required, and every label included with the device has to meet the applicable standards and regulations. When features or hazards are inadequately labeled, misuse can occur and result in harm to the user and a loss of confidence in the device. Good labeling prevents errors and ensures that the user will receive the intended benefits safely.
Labeling is a critical part of the production process for medical device manufacturers. Devices can’t go to market without the required labels. Since regulations cover everything from label design to how they are affixed to a device, manufacturers have to place labeling on equal standing with every other aspect of product quality assurance.
This post will discuss what counts as a medical device label, where they are required, and look at the key points of US and EU medical device labeling regulations. Then we will provide general guidelines for producing labels that will benefit your customers and meet regulatory obligations.
What is a medical device, legally?
The FDA definition of a medical device is something that diagnoses, cures, treats, or prevents a disease or health condition, or otherwise alters the structure or function of the body. A medical device can be any kind of instrument, apparatus, machine, implant, or in vitro reagent; or a part, component, or accessory of another device. Drugs, or anything else that the body metabolizes, are not considered “devices.” Software can be a medical device, as long as it directly serves one of the listed functions. Software that stores data or serves administrative functions does not count.
In Europe, the EU Medical Device Regulation defines medical devices as non-drug products that diagnose, cure, and/or prevent medical conditions and diseases; study anatomical, physiological, or pathological processes for replacement and/or modification purposes; or collect and preserve data samples from human patients.
It may not always be clear to manufacturers within a device’s supply chain whether they are responsible for labeling. Usually, the device manufacturer handles the labels, but sometimes one or more of the following suppliers may also have labeling obligations:
- Re-processors of used devices
- Re-packagers
- Re-labelers
- Kit assemblers
- Specification developers
Manufacturers who are unsure of their labeling requirements should refer to the guidelines provided by their regulatory agency.
What are the requirements for medical device labeling?
In the US and the EU, the requirements for medical device labeling are detailed and extensive, and may be specific to the type of device. Manufacturers should always refer directly to the text of the applicable laws to ensure that they are following the correct procedures and staying compliant.
US FDA Requirements
According to the US FDA, a label is any “display of written, printed, or graphic matter upon the immediate container of any article,” and must be clearly visible even if the “immediate container” has additional packaging. “Label” also includes anything printed on a product container or wrapper, or anything that accompanies the article when it is put up for sale. US courts have found that, by this definition, advertising, posters, brochures, instruction booklets, inserts, and any similar materials are also considered labels for regulatory purposes.
General labeling requirements are defined in CFR Title 21, Part 801. FDA-compliant labels must include the following:
- Manufacturer’s name and business location
- Intended use of the device
- Adequate directions for a layperson to safely operate the device
The label cannot include any false or misleading statements, and must be displayed prominently in an appropriate location. Additionally, the FDA requires medical device manufacturers to use Unique Device Identification labels. UDI allows for the tracking of specific devices, and the required elements must be both printed and machine-legible.

EU MDR Requirements
The EU’s regulations are contained in Annex I, Chapter III of the EU MDR. They are generally aligned with those of the FDA, including the requirement for UDI labeling.
In addition to including labels on or with the product as appropriate, EU manufacturers must publish up-to-date labeling information on their website.
Why is medical device labeling so important?
Many medical devices are designed for use by patients in their homes without direct supervision from a medical professional. Patients, medical professionals, and experienced users all need clear instructions, guidance, and warnings to ensure proper use and safety. When devices are misused or improperly handled, they may fail to function correctly or cause harm to the user. The potential risks for some devices can include serious injury or even death, but even relatively mild errors can negatively impact patient outcomes.
For all stakeholders in the device manufacturing process, nothing should be more important than the health of their end-users. Good labeling practices help to minimize bad experiences across the board. They will also keep you in compliance with the regulatory agency for your region, preventing any costly delays, damaging recalls, or other legal consequences.
General Guidelines for Medical Device Labeling
There isn’t a one-size-fits-all format for a great medical device label. The required elements will vary depending on many factors. It’s up to the manufacturer to ensure that all of the necessary elements are included, and that the label succeeds in conveying the information needed by the patient to have a positive and beneficial experience using the device.
The following tips make up a general outline of best practices for creating effective, compliant labels:
- Start thinking about label design early in your product development process.
- Review your regulator’s guidelines for information that needs to be included in the directions for use.
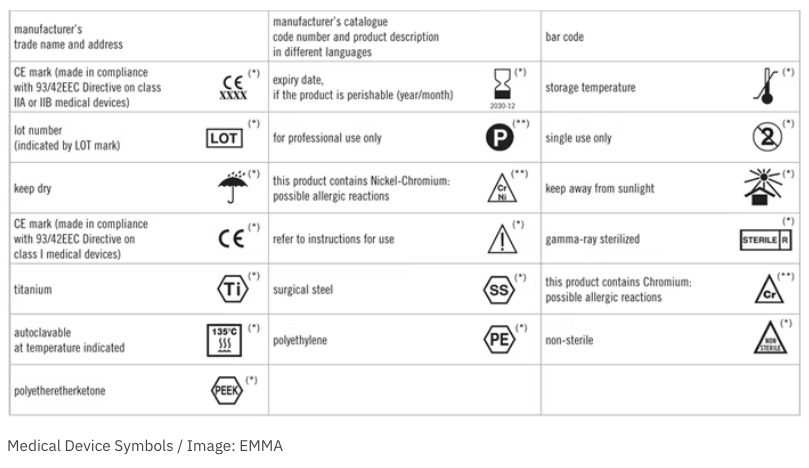
- Use compliant, standardized symbols.
- Use durable materials for labels that will stay attached and legible even after the device has been cleaned according to the instructions.
- Make your label-making processes automated and easily replicable.
- Maintain label templates that can be filled out with required information.
- Reach out to your supply chain partners to ensure that compliance requirements are being met and that none of you are unnecessarily duplicating each other’s labeling responsibilities.
- Engage in usability testing to see if your labels are being interpreted correctly.
- Include sufficient warnings about all known risks, including dangers of off-label use.
Before finalizing your labeling process, you will need to review the law to determine if any additional, specific labeling requirements apply to your business and the type of devices you manufacture.
QMS and Medical Device Labeling
A QMS system designed around the needs of medical device manufacturers can ensure that you’re meeting all of your regulatory obligations while producing labels that uphold the same high quality standards as the devices they come with. The right QMS solution will support your labeling efforts by keeping your manufacturing data up to date, tracking document changes, and ensuring that your processes align with all of the specific regulations that apply to you.