


The Essential Guide to ISO 14971

Medical devices are an irreplaceable part of modern medical care and we depend on medical device manufacturers to produce high-quality, safe, and reliable products. To do that consistently, those manufacturers need a systematic risk management process.
While risk management concerns all manufacturers, medical device manufacturers carry a special responsibility to protect life and health. In the life sciences industry, risk management processes must be a top priority throughout the product’s life cycle.
The manufacture of medical devices is an intensively regulated field and there are numerous standards for risk management in the manufacturing process. The most important of these is the internationally-recognized ISO 14971:2019. In this post, we’ll discuss the ISO 14971 standard, why it’s a good idea for medical device manufacturers to follow it, and look at the ISO 14971 risk management process.
What is ISO 14971?
The International Organization for Standardization created ISO 14971 to assist manufacturers in developing processes for evaluating, monitoring and controlling the risk that their products could cause harm to patients, operators, or other individuals. ISO 14971:2019 is the third and most recent edition of the standard.
The risks that ISO 14971 deals with include both the likelihood that harm may occur and the severity of that potential harm. In a medical device context, risks may be associated with biocompatibility issues, radiation, software security, moving parts, user controls, warning messages, and many other aspects of design and production.
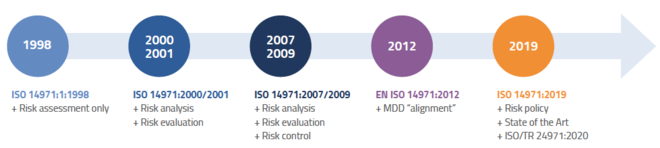
ISO 14971 describes a generic risk management process that covers the product’s entire life cycle from design to disposal. Because ISO 14971 covers a wide range of medical devices, it’s likely most manufacturers will need to apply additional device-specific standards to sufficiently mitigate risk and meet local regulatory requirements.
Compliance with ISO 14971 is not mandatory, and there is no official certification process associated with it. However, it’s considered good manufacturing practice to comply with it, and compliance signifies safety and quality.
What are the benefits of ISO 14971?
The main benefit of ISO 14971 is that it provides a defined framework you can follow to protect the people who use and operate your devices from coming to any harm, which is extremely important from both an ethical and a business standpoint. While ISO 14971 cannot provide specific guidelines for every potential medical device, it focuses on the design, development, and usage issues common to manufacturers in the industry. ISO standards are designed to be compatible with each other, so if you are already following one, you may have an easier time adding ISO 14971 to the mix.
While ISO 14971 is not mandatory in itself, many suppliers, distributors, and customers will be looking for ISO 14971 compliance as a legible sign of a manufacturer’s commitment to safety, security, and rigorous risk management processes. Following a different framework may achieve the same ends, but it might be harder to convey that to others.
ISO 14971 can also help inform business decisions by providing you with a clear assessment of the risks posed by certain products. You may find that certain products are too expensive to develop according to the standards or potentially too hazardous to release.
The ISO 14971 Risk Management Process
ISO 14971 outlines a multi-step process that establishes risk management procedures to be implemented starting from the product’s initial design and carrying forward through its entire life cycle.
Manufacturers should always document the procedures they will be implementing to follow these steps and meet the standard’s requirements. These procedures should be integrated into your QMS.
1. Management Responsibilities
Adequate risk management cannot occur without the support of top management. It is their responsibility to provide resources, training, and authority to the personnel tasked with carrying out risk management procedures.
Top management must also decide on risk acceptability levels. This decision should factor in local regulations, stakeholder input, technological limitations, and whatever other criteria has a bearing on the nature of the device and the conditions it is intended to treat.
2. Risk Management Plan
Risk management procedures should always be planned out in advance. The risk management plan for a medical device should include its criteria for risk acceptability, ways to determine whether risk control measures are successful, and a process for monitoring and responding to emergent risks once the product enters the production and post-production stages.
The manufacturer must also create a risk management file that includes the original risk management plan and other records and documents generated by risk management activities.
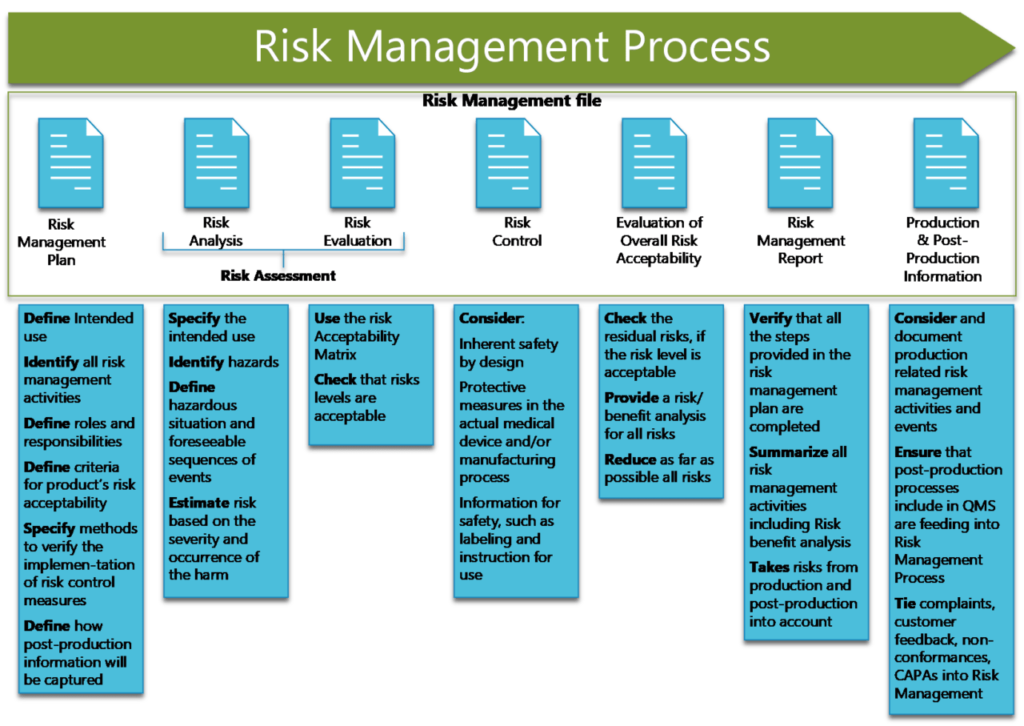
3. Risk Assessment
Risk assessment is a critical part of the risk management process and consists of both analysis and evaluation of risks. There are several steps to follow:
- Document the medical device’s intended use, which should be clearly considered and defined ahead of time. This will help establish the boundaries of “reasonably foreseeable” misuse, which includes both predictable user errors and abnormal uses that can be anticipated, all of which should be addressed by your risk management plan.
- Identify the elements of the medical device that can affect how safe it is. These may include mechanisms, materials, calibration and maintenance requirements, operator skill, and data security.
- Identify the hazards inherent to the medical device and outline some of the predictable events that could lead to the device causing harm.
- Estimate the risk of the hazards identified occurring in terms of both probability and severity.
- Evaluate the risks according to your predefined and documented criteria for risk acceptability. Document the evaluation in the risk management file.
Risks that are found to be acceptable under your criteria are considered to be “residual risks.” Risks that are not acceptable must be mitigated with risk control measures.
4. Risk Control
The most effective way to control risk is to design devices to eliminate it entirely: replacing breakable glass components with soft plastic, for instance.
If risk cannot be completely eliminated at the design level, the next best option is to build additional protective measures into the device. For example, an injection device can be designed such that the needle retracts into the body of the device after use, greatly reducing the chance of accidental puncture wounds.
When the second option is not feasible, instructions and warnings can be included with the device to educate users about how to avoid hazardous situations.
5. Overall Residual Risk Evaluation
Once all identified risks have been mitigated to acceptable levels, the remaining residual risks must be evaluated. The evaluation process, and the acceptability criteria for all residual risks combined, should be documented in the risk management plan. After this evaluation, the manufacturer should disclose to customers any residual risks that were deemed acceptable.
6. Risk Management Review
Once the product has been developed — but before it ships — the risk management plan needs to be reviewed to ensure that it was comprehensive and properly executed. Review findings should be reported and added to the risk management file. This file serves as documentary proof that you have established adequate risk management objectives and met them.
7. Production and Post-Production
In ISO 14971:2019, there are four steps outlined in the production and post-production phase:
- Establish a system for collecting and reviewing production and post-production information relevant to device safety.
- Actively collect relevant data from users, industry partners, suppliers, and other sources.
- Review the information to determine whether new risks can be identified, if previously identified risks are no longer within acceptable levels, if the benefits no longer outweigh the residual risks, or if other circumstances related to risk have changed.
- If unacceptable risks have been uncovered in step 3, the manufacturer must take appropriate actions (as outlined in the standard) to bring risks back under control.
Support Risk Management with QMS
Risk management is a complex and comprehensive process, especially where people’s lives hang in the balance. Making the best possible effort to ensure that their products won’t cause harm to patients or providers is an obligation of all medical device manufacturers. The ISO 14971 standard provides a comprehensive, objective framework for meeting this great responsibility.
While it may not be required as part of the standard, a QMS can make ISO 14971 much easier to manage, track, and implement. The right solution can help you calculate risk scores, and align risk management with design and development activities. A powerful QMS will provide key insights into the actions you can take to lower the probability of risk –maximizing the positive and beneficial experiences customers will have with your products. Start setting your eQMS foundations today.